肢骨纹状肥大
病因学
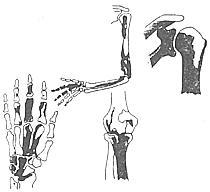 是一种先天性发育畸形,较罕见。无遗传性及家族史。发病从儿童开始,男女相等。大都侵犯单肢,下肢多见于上肢。
是一种先天性发育畸形,较罕见。无遗传性及家族史。发病从儿童开始,男女相等。大都侵犯单肢,下肢多见于上肢。
本病病因不明,认为多系先天发育畸形,为一种骨膜下毛细血管扩张所致的骨膜骨发育异常,文献中曾有与家族性脆骨性骨硬化伴发的报道,故认为其可能是引起孤立性家族性脆骨症基因部位的二次突变所致。
病理改变
症状表现
(一)疼痛最常见,约占1/2,而且年龄越大,疼痛越明显,一般为隐痛或钝痛,活动时加重。
(二)患肢关节活动受限。这是由于关节周围骨质增生,及软组织内骨沉积所致,很少是因为关节面被破坏所致。
检查
X线表现:好发于四肢长骨,以单肢多见。在骨干的一侧有不规则的骨质增生。骨干的外形逐被破坏,犹如在燃烧中的蜡烛的熔蜡从旁边流下。增生的骨无结构,骨骺及短骨常表现为斑点状,可以超越过关节侵犯远端的骨质,但不侵犯关节面。骨盆及肩胛骨亦表现为密度增加及有斑点、颅骨、脊柱及肋骨少见。
鉴别诊断
并发症
治疗措施
由于本病是一慢性疾病,只在发育期病情进展迅速,成年后缓慢,且不影响生命,但本病可出现关节运动受限、关节畸形等。目前无特殊疗法,可用物理疗法和对症治疗以减轻痛苦,预后较好。对于疼痛剧烈、严重影响关节活动者,可考虑手术治疗,如清除钙化的软组织、肌腱延长术、筋膜切开与关节囊切开术、矫正性截骨术、交感神经切断术、甚至截肢。
预后
附件列表
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。