小儿混合性结缔组织病
病因
(一)发病原因
MCTD病因未明。其发病可能与免疫紊乱有关,有关证据为血清中持续存在高滴度抗RNP抗体,显著多克隆高丙球蛋白血症,B淋巴细胞活性过高,抑制性T细胞缺陷,疾病活动期血循环免疫复合物增加,25%病人有低补体血症,发现血管壁、肌纤维膜、肾小球基底膜及皮肤表皮和真皮结合部位有IgG、IgM、IgA和补体沉积。本病与HLA之间的关系正在研究中,有人认为其有特定免疫遗传学背景,与HLA-DR4、-DR5密切相关,认为MCTD为一独立性疾病。
(二)发病机制
MCTD的发病机制尚不清楚。以下证据表明,其发病机制可能与免疫介导有关:循环免疫复合物在肾小球沉积,血清抗RNP抗体阳性,许多组织广泛的淋巴细胞、浆细胞的浸润,高gamma;-球蛋白血症及T细胞抑制。抗RNP抗体的抗原位点是核RNA蛋白复合物,低分子量小核糖核酸蛋白(SnRNPs)参与核RNA合成。MCTD患者血清中除SnRNPs抗体外,还有高分子量与核基质连接的其他抗体。已经证实IgG抗RNP抗体通过Fc受体与细胞内部连接,但通过何种途径激活免疫反应仍不清楚。
SLE患者的血清抗体与MCTD不同,两者异常T淋巴细胞亚群也各不相同。MCTD免疫复合物数量增加可能与网状内皮系统免疫复合物清除受损有关。
遗传因素可能与MCTD有关,但是MCTD和SLE有不同的家族发病体系。
症状
各种结缔组织病虽然在小儿乃至老年期均可发病,但多见于成人,小儿较少。发病年龄从5岁至80岁,平均37岁,80%病人为女性。发生在儿童期的有风湿热、儿童类风湿性关节炎、强直性脊柱炎、系统性红斑狼疮、血管炎、过敏性紫癜、皮肤粘膜淋巴结综合症、婴儿多动脉炎、皮肌炎,硬皮病等。
早期临床表现
早期症状轻微,如雷诺现象、肌肉痛、关节痛、疲劳,检查高球蛋白血症,抗核抗体可疑阳性,经数月或数年后进展为典型表现而确诊。雷诺现象、手及手指皮肤水肿增厚的现象可在其他表现出现前数月或数年显示。肌无力(有或无疼痛或触痛)可拟诊为肌炎。胸膜炎、心包炎亦可为早期征象,如合并发热、关节痛、红斑性皮疹,可诊断为SLE。小儿早期肾脏、心脏受累及血小板减少症在本病中早期较成人为多见。早期也可为呼吸困难的肺部受累表现,及淋巴腺病拟诊为淋巴瘤者。
临床主要表现:
1.皮肤病变:
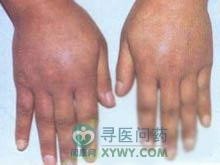
几乎每个MCTD患者都有皮肤受累。最常见的是手肿胀,手肿胀占66%~88%,特别是手指呈锥形头尖腊肠样,皮肤绷紧、增厚并伴显著水肿,这是皮肤胶原增加所致。但不伴明显硬化,肿胀不过腕,可合并毛细血管扩张征。狼疮样皮疹包括急性两颊皮疹;弥漫性,非痂皮性,红斑性病损;及(或)慢性盘状皮肤病损;网状青斑。其他包括指节萎缩性红斑皮肌炎相似的上眼睑有紫红斑、播散性非疤痕性脱发及色素异常,面和手脱屑性毛细血管扩张症和指甲周围的毛细血管扩张症。大约3/4患者有硬皮病样皮肤改变。
2.雷诺现象:
达90%~100%,肢端接连出现苍白、发组和潮红三相反应,多发生于上肢,两侧对称,也可累及下肢,或同时波及上下肢,偶尔发生于耳朵、鼻端、颊部或领部。患儿对凉水及寒冷反应明显。少数重症患儿可发生指、趾端缺血性溃疡或坏死。
3.关节病变:
占87%~100%的病人有显著关节炎,多关节痛。关节一般无畸形,有时偶见如类风湿性关节炎的畸形,少数有关节糜烂侵蚀、畸形。但多限制在手、腕或足部,可伴有皮下结节。
4.肺部表现:
有些临床上可无呼吸道症状,无症状患者70%已有肺功能异常和(或)X线检查异常。可伴少量胸腔积液,呼吸困难,约2/3患者有肺弥散功能减退,1/2病人有限制性通气功能障碍,少数胸膜炎,非间质纤维化,肺动脉高压。肺功能测定3/4病例受损,最常见为一氧化碳弥散功能障碍和肺活量减低,呼吸受限,某些有运动性呼吸困难和肺动脉高压,后者可继发于肺纤维化或肺小动脉内膜增生。X线摄片可见肺基底部网状结节阴影、变换位置的肺实质变化。
5.消化道表现:
消化道累及占70%,可见食管扩张、食管远端2/3蠕动减弱,可出现进食后发噎和吞咽困难。十二指肠扩大和大肠憩室,曾报导有肠壁囊样积气症。肠道功能障碍致痛性痉挛、胀气、便秘和腹泻交替及吸收不良。也可有广泛胃肠道病变。
6.心脏表现:
小儿心脏受累较成人多见,心脏病变约30%有心包炎,尚可有心肌炎、心律失常、心功能不全、完全性传导阻滞、节律紊乱和心力衰竭,亦可有瓣膜病变如二尖瓣闭锁不全和狭窄,曾报导一例主动脉瓣闭锁不全引起心力衰竭。
7.肾脏表现:
肾脏累及少,约5%~25%,肾穿刺示弥漫性膜性增生性改变、弥漫性膜性肾炎、局灶性肾小球肾炎、肾小球血管膜细胞增生、细胞浸润、内膜增殖和血管闭塞。肾损害表现为蛋白尿和血尿,28%病人有血尿、蛋白尿、管型尿,严重肾功能衰竭并可因进行性肾功能衰竭死亡。
8.神经病变:
约占10%,最常见三叉神经痛,此外有多发性神经炎、无菌性脑膜炎、癫痫、横贯性脊髓炎、马尾综合征、发作性血管性头痛和精神病。
9.血液系统:
有中等度贫血、白细胞减少,Coombe阳性溶血性贫血和血小板减少症较少见,严重的血小板减少者需脾切除,可因颅内出血死亡。
10.其他:
约30%病例有肝脾肿大和浅表淋巴结肿大。严重肝功能异常者罕见。此外,可伴同干燥综合征(7%~50%)或桥本甲状腺炎(6%)。
检查
1.外周血象
常有中度贫血,血白细胞 <4×109/L,偶见血小板减少。血沉增高。
2.X线检查
30%的患者胸片示不规则小片状阴影,为肺部增殖性血管病变所致。25%~60%的病例关节有侵袭性病变。
3.自身抗体检测
(1)抗核抗体(ANA)免疫荧光检测显示强阳性斑点型ANA。
(2)抗RNP抗体几乎100%的患者抗RNP抗体阳性(血凝法>1∶1 000),抗Sm抗体阳性率低于10%。Sm及RNP抗原均属核内小核糖核蛋白(snRNPs)。SnRNPs是由U族小分子RNP(UsnRNA)与核糖核蛋白组成的复合物。哺乳动物的UsnRNP至少有13种以上(U1~U13)。抗RNP抗体仅能沉淀U1,故又叫抗U1RNP抗体。抗Sm抗体则能沉淀U1,U2及U4~U65种UsnRNP。抗U1RNA抗体与病情活动有关,其中100%抗snRNP-70kD抗体阳性,85%抗snRNP-A多肽抗体阳性。有人认为抗U1RNA抗体是MCTD的标志性抗体,但 SLE及干燥综合征患者血清抗U1RNP抗体也可阳性。
(3)抗hnRNP抗体hnRNPs由近30种不同蛋白组成,能与mRNA前体hnRNA结合,参与mRNA前体剪切。抗hnRNP-A2/RA33抗体针对的抗原为hnRNPs蛋白A2、B1、B3(称为RA33复合体)。MCED的hnRNP-A2/RA33抗体阳性率为35%~40%,常伴抗Sm阴性。
(4)抗dsDNA抗体阳性率仅24%。
诊断
目前尚无MCTD的诊断标准,Sharp(美国)于1986年在日本召开的混合性结缔组织病和抗核抗体的学术讨论会上提出的MCTD的诊断标准(成人)可供参考。
1.主要标准
①肌炎(重度);
②肺部累及:包括CO2弥散功能<70%,肺动脉高压,肺活检示增殖性血管损伤;
③雷诺现象或食管蠕动功能降低;
⑤抗ENA,1∶1000,抗U1RNP抗体(+)及抗Sm抗体(-)。
2.次要标准
①脱发;
②血白细胞减少或<4×109/L;
③贫血;
④胸膜炎;
⑤心包炎;
⑥关节炎;
⑦三叉神经病变;
⑧颊部红斑;
⑩肌炎(轻度);
3.确诊
4个主要指标加上抗RNP抗体(+),滴度>1∶4000,且抗Sm抗体(-)。
4.可能诊断
3个主要指标或2个主要指标加上抗RNP抗体阳性,滴度>1∶1000。
5.可疑诊断
3个主要指标,抗RNP可为阴性;或2个主要指标加上抗RNP(+),滴度1∶100;或1个主要指标加上3个次要指标,加上抗RNP(+),滴度1∶100。
诊断鉴别
1.重叠综合征(overlappingsyndrome)是指同一个患者同时或先后患有明确2种或2种以上结缔组织病的重叠,因此与MCTD明显差别。尽管MCTD临床上有多种病的重叠症状,但它有其自身的诊断标准和特点。
重叠综合征临床表现为同一时期内并存两种以上风湿性疾病,如系统性红斑狼疮与结节性多动脉炎并存,系统性红斑狼疮与系统性硬化症并存,幼年型类风湿性关节炎除有严重关节炎和皮下小结外,还有抗核抗体阳性和血管炎,即与系统性红斑狼疮并存。此外,系统性硬化症也可出现抗核抗体阳性、严重关节炎、白细胞减少、溶血性贫血等。
重叠综合征也可先后出现两种以上风湿性疾病,如患皮肌炎多年后,又出现双手指关节炎,可视为皮肌炎与类风湿关节炎重叠。系统性红斑狼疮的病程进展过程中,又出现了典型的类风湿关节炎的关节畸形,可诊断为系统性红斑狼疮和类风湿关节炎的重叠。
重叠综合征的治疗较治疗单纯患某一种疾病困难。应用药物应取决于以何种风湿性疾病为主。主要药物为肾上腺皮质激素和免疫制剂,剂量和用法参照各有关风湿性疾病节。
本综合征的预后较单一的风湿性疾病要差。同时还取决于哪两种病的重叠。与MCTD相比,MCTD的5年存活率为93%,而重叠综合征的存活率仅为53%。
并发症
死亡原因为肺部疾病、肾功能衰竭、心肌炎、心肌梗死、心力衰竭、脑栓塞、脑出血和泛发性血管炎、结肠穿孔。儿童的混合性结缔组织病病情较严重,中枢神经系统、心和肾累及较成人为多,关节炎亦常见,可有严重血小板低下。可发生指、趾端缺血性溃疡或坏死;少数并发胸膜炎,非间质纤维化,肺动脉高压;可并发心包炎、心肌炎、心律失常、心瓣膜病变、心功能不全等;可并发多发性神经炎,无菌性脑膜炎,癫痫等。尚有淋巴结肿大,少数肝脾肿大。
治疗
MCTD仅有关节炎时可用非甾体类激素如布洛芬、萘普生或阿司匹林或小剂量激素(参考类风湿性关节炎的治疗);皮肤损害可用抗疟药如氯喹,也可用泼尼松。如出现重要脏器受累,明显的浆膜炎、肌炎及进行性肺或食管病变时应使用大剂量糖皮质激素治疗,但对间质性或限制性肺病变疗效不确切。肾受累则以糖皮质激素加环磷酰胺静脉冲击治疗为宜(具体可参考SLE治疗)。
该病预后与SLE相仿,但较硬皮病好。6~12年的病死率为13%,主要死于严重的心肺并发症如肺动脉高压、肺心病、心力衰竭和心包填塞,也可死于肠穿孔或败血症。
预后
儿童的混合性结缔组织病病情较严重,病程较长,但经适当治疗可获临床缓解。该病预后与SLE相仿,但较硬皮病好。6~12年的病死率为13%,主要死于严重的心肺并发症如肺动脉高压、肺心病、心力衰竭和心包填塞,也可死于肠穿孔或败血症。
预防
本病死因主要是感染等并发症及激素治疗的不良反应,极少数严重病例可死于肾功能衰竭和中枢神经系统病变,故对本病的早期诊断和合理有效治疗可预防并发症的发生和延长存活期。早期加强保暖,避免外伤,内服皮质激素和活血通络的药物,及时到医院检查治疗。忌寒冷、潮湿刺激。结缔组织病患者在寒冷、潮湿环境下,经常会感觉到关节疼痛加剧,出现雷诺现象或者两手足出现瘀点、疼痛加剧等。
饮食
附件列表
词条内容仅供参考,如果您需要解决具体问题
(尤其在法律、医学等领域),建议您咨询相关领域专业人士。